
 |
 |
 |
 |
 |
Artigo
|
|
| Determinação do flavonoide quercetina em extratos de planta por infravermelho (ATR-FTIR) e quimiometria Determination of the flavonoid quercetin in plant extracts by infrared spectroscopy (ATR-FTIR) and chemometrics |
|
Nadia B. da CostaI,* I. Departamento de Química, Instituto Federal de Educação, Ciência e Tecnologia do Maranhão, Campus Monte Castelo, 65030-005 São Luís - MA, Brasil Recebido: 21/12/2024 *e-mail: nadya152009@hotmail.com This work evaluated mid-infrared spectroscopy combined with chemometrics for qualitative and quantitative analysis of flavonoids in plant extracts. The exploratory data analysis was conducted using principal component analysis (PCA) and hierarchical cluster analysis (HCA), while the calibration model was developed using the partial least squares (PLS) regression technique. Methanolic extracts were obtained from 15 plant species collected during rainy and dry seasons, totaling 26 samples. Ultraviolet-visible (UV-Vis) spectroscopy was used as a reference method for the determination of flavonoids. PCA and HCA grouped the samples according to their chemical composition, with no significant variations due to seasonality. The extract of "quebra-pedra" (Phyllanthus niruri) formed a distinct cluster related to the spectral bands at 1012 and 1047 cm-1, attributed to C-O stretching in carbohydrates and cellulose derivatives. The regression model presented a root mean square error of calibration (RMSEC) of 0.06, root mean square error of cross-validation (RMSECV) of 0.13, root means square error of prediction (RMSEP) of 0.20, and residual predictive deviation (RDP) of 1.62. These results demonstrate that the combination of Fourier transform infrared spectroscopy (FTIR) and PLS presents itself as an alternative to predicting the concentration of flavonoids in complex samples, such as plant extracts. INTRODUÇÃO Estudos têm demonstrado que muitas plantas possuem componentes químicos com diversas atividades biológicas.1-3 Dentre os constituintes bioativos mais comuns e amplamente distribuídos nas plantas, destacam-se os flavonoides.4 Os flavonoides pertencem a uma classe de compostos fenólicos que apresentam 15 átomos de carbono em sua molécula, organizados em uma configuração C6-C3-C6. Entre as várias atividades biológicas atribuídas aos flavonoides, destacam-se propriedades antioxidantes, antibacterianas, anti-inflamatórias, antivirais, entre outras.5 Técnicas analíticas, como a espectrometria ultravioleta-visível (UV-Vis), a cromatografia gasosa e a espectrometria de massa, têm sido amplamente utilizadas para determinar a concentração de flavonoides.6 No entanto, esses métodos frequentemente envolvem procedimentos laboriosos, etapas de extração demoradas e o uso de reagentes como solventes orgânicos (por exemplo, metanol, acetonitrila e hexano), alguns dos quais são tóxicos e apresentam alto custo. A espectroscopia de infravermelho (IR) é uma ferramenta bem estabelecida em química analítica, proporcionando um método não invasivo, não destrutivo e rápido para avaliar a composição química de uma ampla gama de amostras.7 Essa técnica requer apenas pequenas quantidades de amostra e permite a análise de diversos tipos de matrizes, incluindo sólidos, pós, filmes, líquidos e gases.8 No entanto, seus principais desafios incluem a interpretação dos espectros de misturas complexas e a necessidade de desenvolver e manter modelos de quantificação robustos para análises quantitativas.7 Nesse contexto, o uso de métodos quimiométricos torna-se essencial para interpretar as informações espectrais, melhorar a seletividade da técnica e possibilitar uma análise multicomponente rápida e eficiente.9 Métodos quimiométricos são ferramentas matemáticas e estatísticas aplicadas à química, utilizadas para planejar experimentos, analisar dados químicos e interpretar resultados de forma eficiente e precisa. Esses métodos são especialmente valiosos para extrair informações relevantes de conjuntos de dados complexos e multidimensionais, como os gerados em análises espectroscópicas.10 Ferramentas quimiométricas bem estabelecidas, como a análise de componentes principais (PCA) e a análise hierárquica de agrupamento (HCA), são amplamente empregadas na análise exploratória de dados de espectroscopia no infravermelho.11 PCA é uma técnica não supervisionada, rápida e confiável, utilizada para reduzir a dimensionalidade de conjuntos de dados multivariados complexos. Sua simplicidade permite que diferenças espectrais sejam apresentadas de forma intuitiva.12 A PCA transforma os dados originais em um espaço de menor dimensão, baseado em um novo conjunto de eixos denominados componentes principais (PCs). HCA é outra técnica não supervisionada de reconhecimento de padrões, adequado para descobrir padrões naturais de comportamento entre amostras com base em seu perfil multivariado. HCA é útil para reduzir a dimensionalidade dos dados multivariados agrupando-os de forma hierárquica. Os resultados são apresentados na forma de uma árvore conhecida como dendrograma, que nada mais é do que um gráfico que representa a estrutura hierárquica dos dados, em que os comprimentos dos ramos da árvore representam o grau de similaridade entre os objetos.13 Embora o reconhecimento de padrões seja útil para explorar e identificar tendências nos dados, para uma análise quantitativa é necessário o uso de métodos de calibração multivariada. Dentre esses métodos, o mais utilizado é a regressão por mínimos quadrados parciais (PLS).14 PLS correlaciona a matriz de medida X e a matriz de resposta Y e, simultaneamente, as decompõe considerando a relação entre elas. O PLS encontra o melhor modelo de correção linear projetando X e Y para novos espaços de fatores.15 Vários estudos da literatura16-18 sugerem que a espectroscopia de infravermelho médio (MIR) combinada com quimiometria é eficaz para análise de alimentos e plantas em termos de autenticação, detecção de adulteração, determinação da composição química e avaliação de qualidade. No presente estudo, foi determinado o teor de flavonoides em 15 espécies de plantas do cerrado maranhense, coletadas em diferentes períodos do ano, utilizando a espectroscopia no infravermelho com transformada de Fourier por reflectância total atenuada (FTIR-ATR) associada à quimiometria. Para a análise exploratória dos dados, foram empregados os técnicas HCA e PCA, enquanto a quantificação da concentração de flavonoides foi realizada por meio de PLS. Os resultados demonstraram que é possível determinar o conteúdo de flavonoides de maneira rápida e eficiente, com mínimo preparo de amostra, utilizando FTIR-ATR. Além disso, este trabalho fornece uma base de dados para a avaliação da impressão digital espectral desses extratos de plantas, coletados em diferentes épocas do ano, contribuindo para estudos futuros sobre a variabilidade sazonal e a composição química de espécies vegetais.
PARTE EXPERIMENTAL Coleta das amostras As folhas de 15 espécies vegetais do cerrado maranhense foram coletadas em diferentes períodos do ano, abrangendo tanto o período chuvoso quanto o período de estiagem, totalizando 26 amostras. As coletas foram realizadas no município de Buriti, Maranhão, localizado na região Nordeste do Brasil, nas coordenadas geográficas 3º56'24" S de latitude e 42º55'05" W de longitude. As espécies vegetais coletadas estão detalhadas na Tabela 1.
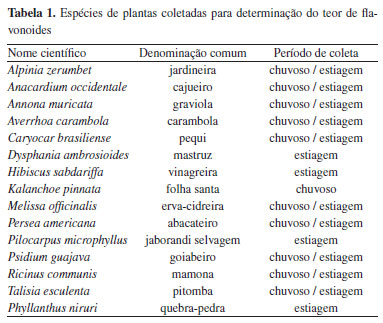
As folhas das espécies vegetais foram acondicionadas em frascos plásticos e transportadas para o laboratório de química para posterior análise. As coletas foram realizadas em duas etapas, correspondentes ao período chuvoso e ao período de estiagem, conforme especificado na Tabela 1. A identificação botânica das plantas foi conduzida no Herbário do Maranhão (MAR) da Universidade Federal do Maranhão (UFMA), utilizando exsicatas preparadas a partir das amostras coletadas. Obtenção dos extratos As folhas das plantas foram inicialmente secas em estufa com circulação de ar por um período de 7 dias a 45 ºC. Após a secagem, as folhas foram trituradas em moinho para obtenção de um pó homogêneo. Para a extração, 20 g do pó das amostras foram misturados com 200 mL de metanol e submetidos a ultrassom por 1 h. Em seguida, o solvente foi separado do material insolúvel por filtração, e o extrato líquido obtido foi concentrado em um evaporador rotativo a aproximadamente 30 ºC. O extrato foi concentrado até a completa evaporação do solvente, resultando em resíduo seco. Não foi adotado um volume final fixo. Para determinação do rendimento dos extratos, os recipientes foram previamente pesados (sem tampa) antes da adição das amostras. Após a evaporação completa do metanol, os frascos contendo os extratos secos foram novamente pesados, e a massa obtida foi considerada como massa do extrato seco. O rendimento foi calculado com base na massa do extrato seco em relação à massa da amostra vegetal inicial (20 g), utilizando rendimento (%) = (massa do extrato seco / massa da amostra vegetal seca) × 100. Determinação da concentração de flavonoide A concentração de flavonoide foi determinada pelo método cloreto de alumínio (AlCl3) usando a quercetina (≥ 95%, grau HPLC, sólida, Sigma-Aldrich), como composto de referência.5 Inicialmente, foi preparada uma solução estoque dos extratos vegetais na concentração de 10 mg mL-1, utilizando metanol como solvente. Para a análise, foram transferidos 0,5 mL da solução estoque para um tubo de ensaio, seguidos da adição de 3,0 mL de metanol (Merck); 0,2 mL de AlCl3 (Isofar) a 10% (m/v) e 0,2 mL de solução de acetato de potássio (CH3CO2K, Isofar)1 mol L-1, 0,2 mL de uma solução aquosa de cloreto de alumínio a 10% (m/v) e 0,2 mL de uma solução de acetato de potássio 1 mol L-1. A mistura foi então diluída com água destilada até completar o volume de 10 mL. Após incubação por 30 min, a absorbância da solução foi medida a 428 nm utilizando um espectrofotômetro UV-Vis (Biospectro SP-22). A amostra em branco foi preparada substituindo-se o extrato vegetal por metanol, mantendo-se os demais reagentes e volumes. Para a construção da curva analítica, soluções-padrão foram preparadas a partir de uma solução-estoque de quercetina certificada, com concentrações variando de 2 a 20 µg L-1, contendo as mesmas proporções de AlCl3 e CH3CO2K das amostras analisadas. Cada ponto da curva foi analisado em triplicata, sendo utilizado o valor médio das absorbâncias para a construção da curva de calibração. Os resultados foram expressos em miligramas de equivalentes de quercetina por grama de extrato seco (mgQE g-1). Obtenção dos espectros de infravermelho (FTIR) Os espectros das amostras foram obtidos utilizando um espectrômetro Shimadzu IRAffinity-1 FTIR, acoplado a um acessório de reflexão total atenuada (ATR) com cristal de seleneto de zinco (ZnSe). Os dados foram processados com o software IRsolution 1.6SU1, que gerou os espectros de cada amostra com seus respectivos valores de absorbância. As análises foram realizadas no intervalo espectral de 4000 a 650 cm-1, com 64 varreduras por espectro e uma resolução de 4 cm-1. Antes de cada análise, foi registrado um espectro do branco, utilizando o ar ambiente como referência, para compensar as contribuições de absorção atmosférica, especialmente de vapor d'água e dióxido de carbono. Calibração por PLS e análise exploratória por PCA e HCA A análise quimiométrica foi realizada utilizando o software The Unscrambler® X ,versão 10.4, 2016 (Camo Analytics, Oslo, Noruega). Inicialmente, os espectros foram submetidos à suavização por média móvel, seguida da aplicação da primeira derivada utilizando o filtro de Savitzky-Golay com uma janela de 15 pontos. Em seguida, os dados foram centralizados na média antes da aplicação da PCA e PLS. Métodos não supervisionados de reconhecimento de padrões, como Análise de componentes principais (PCA) e análise hierárquica de agrupamento (HCA), foram empregados para análise multivariada do conjunto de dados. A regressão por mínimos quadrados parciais (PLS) foi utilizada para construir modelos matemáticos empíricos que correlacionassem a variável de interesse com os sinais analíticos, possibilitando a quantificação do analito. O critério utilizado para a divisão entre os conjuntos de calibração e validação seguiu a proporção de aproximadamente 70% das amostras para o modelo de calibração e 30% para a validação externa, conforme recomendado por Ferreira.13 O desempenho dos modelos foi avaliado por meio do erro quadrático médio da calibração (RMSEC), do erro quadrático médio da validação cruzada (RMSECV), do erro quadrático médio da predição (RMSEP), do coeficiente de determinação (R2) e do desvio padrão residual (RSD).
RESULTADOS E DISCUSSÃO Rendimento dos extratos Na Figura 1 são mostradas as espécies vegetais e seus respectivos rendimentos de extratos metanólicos. Devido às restrições logísticas de deslocamento durante o período de estiagem, algumas plantas não puderam ser coletadas.
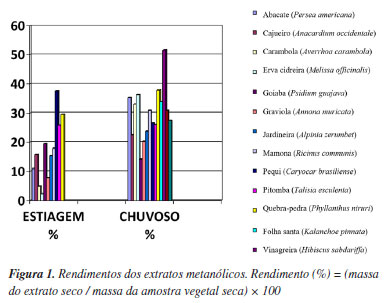
Dentre as espécies analisadas, quebra-pedra (Phyllanthus niruri) e pequi (Caryocar brasiliense) foram as que apresentaram maiores rendimentos para o período de estiagem, 29,55 e 37,5%, respectivamente. Enquanto que, para o período chuvoso, os maiores rendimentos foram observados para vinagreira (Hibiscus sabdariffa) (51,6%) e quebra-pedra (Phyllanthus niruri) (37,8%). De um modo geral os maiores rendimentos foram obtidos para os extratos das folhas coletadas no período chuvoso, o que sugere que ambiente chuvoso, quente e úmido favoreve uma maior disponibilidade de substâncias a serem extraídas. Esse resultado está de acordo com o observado por Maharaj et al.,19 em pesquisa desenvolvida na África do Sul onde o aumento no rendimento dos extratos metanólicos foi observado para folhas coletadas em períodos mais chuvosos e quentes. Todas as amostras foram submetidas ao mesmo método de extração, no entanto, por se tratarem de espécies vegetais distintas, estas apresentam concentrações variadas de compostos solúveis em metanol, como alcaloides, flavonoides, e taninos, o que pode afetar o rendimento. Zilli et al.20 investigaram a influência da sazonalidade sobre o rendimento de extratos secos de diferentes amostras de Ilexparaguariensis (erva-mate). Os resultados indicaram que a época do ano em que as folhas foram coletadas afetou significativamente o rendimento dos extratos, sugerindo que fatores sazonais, possivelmente relacionados a variações climáticas, influenciam a composição química e o rendimento dos extratos. Detecção de flavonoides por UV-Vis A Figura 2 mostra o resultado da concentração de flavonoides equivalentes em quercetina, nos extratos de diferentes espécies vegetais coletadas em períodos de chuva e estiagem.
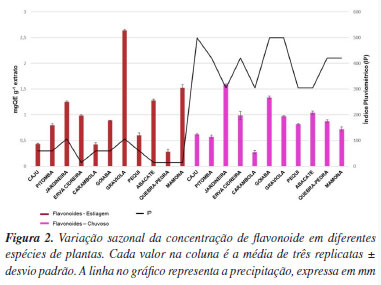
Obseva-se que a maior concentração de flavonoide foi obtida para graviola (Annona muricata) no período de estiagem. Segundo Pourcel et al.,21 compostos fenólicos, como taninos e flavonoides, são responsáveis pela defesa antioxidante da planta quando esta sofre estresse ambiental ou quando passa por diferentes estágios morfológicos de desenvolvimento. Extratos obtidos de plantas coletadas no período seco apresentaram o maior valor médio de flavonoide (1,01 ± 0,6) em comparação ao valor médio para o período chuvoso (0,88 ± 0,36). Para comparação das médias foi aplicado um teste t de Student, onde concluiu-se que os dois grupos (chuvoso e estiagem) eram estatisticamente semelhantes. Dessa forma, não houve diferença estatística entre os resultados (p > 0.05), demonstrando que o fator sazonalidade não influenciou nas concentrações de flavonoides obtidos para este grupo de amostras. Como foi verificado na Figura 2, a quantidade total de extrato foi influenciada pelas condições de coleta (período chuvoso), mas a concentração de flavonoides não apresenta variação significativa entre os períodos conforme o teste t de Student. Dessa forma, a fim de obter resultados mais robustos, seria necessário aumentar o número de amostras coletadas nos dois períodos, bem como considerar outros fatores ambientais, como incidência de luz e temperatura, que podem influenciar no acúmulo de compostos bioativos. Espectros de infravermelho (FTIR-ATR) dos extratos de vegetais Os espectros de FTIR dos extratos de vegetais e do padrão de quercetina são apresentados nas Figuras 3a e 3b. Para reduzir o espalhamento e o ruído, os espectros foram pré-processados com correção de linha base e alisamento (smoothing) por média móvel com janela de 7 pontos, conforme mostrado na Figura 3a. Após a aplicação do pré-processamento foi possível observar uma redução no espalhamento dos espectros e no ruído.
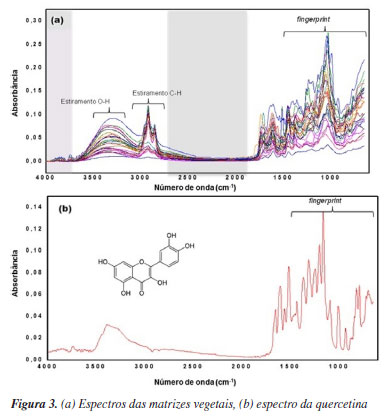
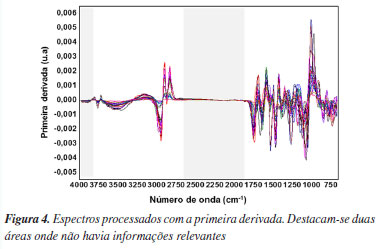
O espectro de absorção no infravermelho de cada composto possui um padrão único conhecido como impressão digital (fingerprint), localizada entre 1400 a 400 cm-1.22 Tanto nos espectros do padrão de quercetina (Figura 3b) quanto nos espectros dos extratos vegetais, observa-se que as maiores informações estão localizadas nessa região. No espectro padrão de quercetina (Figura 3b), o estiramento dos grupos OH foi detectado em 3406 e 3283 cm-1, enquanto a deformação angular OH da função fenol foi observada em 1379 cm-1. A banda em 1666 cm-1 foi atribuída ao estiramento do grupo aril cetona (C=O), e as bandas de estiramento do anel aromático (C=C) foram localizadas em 1610, 1560 e 1510 cm-1. Adicionalmente, a banda de deformação angular no plano de C-H em hidrocarbonetos aromáticos foi observada em 1317 cm-1, e as bandas de deformação fora do plano foram evidentes em 933, 820, 679 e 600 cm-1. As bandas em 1263, 1200 e 1165 cm-1 foram atribuídas ao estiramento C-O no anel do éter arílico, ao estiramento C-O no fenol e ao estiramento e deformação C-CO-C na cetona, respectivamente. Uma diferença perceptível entre os espectros do padrão de quercetina e dos extratos vegetais foi a presença de bandas entre 2800 e 2900 cm-1 nos espectros dos extratos, que estão ausentes no espectro da quercetina. Essas bandas correspondem às vibrações de estiramento C-H, específicas para os grupos CH3 e CH2 encontrados em lipídios.23 Essa característica indica a presença de compostos lipídicos nos extratos vegetais, que não estão presentes no padrão de quercetina. Por outro lado, a semelhança entre os espectros dos extratos vegetais e o padrão de quercetina, especialmente na regiãode impressão digital, sugere a presença de compostos com estruturas similares nos extratos analisados. Análise exploratória dos dados Embora os espectros originais tratados com correção de linha base e alisamento tenham reduzido o espalhamento e o ruído do sinal, os melhores resultados para as análises quimiométricas foram obtidos após a aplicação da primeira derivada de Savitzky-Golay, utilizando um polinômio de segunda ordem e uma janela de 15 pontos. Além disso, os dados foram centralizados na média, o que desloca as coordenadas para o centro do conjunto de dados. Esse procedimento permite destacar diferenças nas intensidades relativas das variáveis, facilitando a análise multivariada.24 A Figura 4 apresenta os espectros processados com a primeira derivada, destacando as regiões excluídas devido à ausência de informações espectrais relevantes.
A análise de componentes principais (PCA) foi realizada utilizando os dados processados com a primeira derivada. Os gráficos de escores foram construídos com base nos períodos sazonais e nas diferentes espécies de plantas, revelando a formação de 3 grupos distintos (Figura 5). De acordo com o gráfico de escores, duas componentes principais explicaram 71% da variância dos dados. Segundo Ferreira13 a variância total explicada em uma PCA deve estar entre 70 e 95% para capturar adequadamente a maior parte da informação original dos dados.
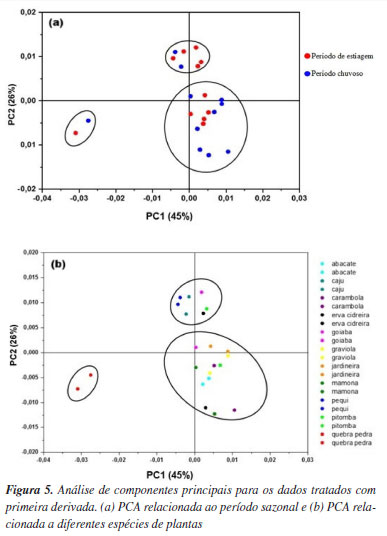
As folhas utilizadas no preparo dos extratos foram coletadas nos períodos chuvoso e de estiagem. Teoricamente, esperava-se uma separação mais clara das amostras de acordo com a sazonalidade. No entanto, observa-se que, em alguns casos, como nas amostras de erva-cidreira (Melissa officinalis) (destacadas em amarelo), há uma tendência de separação entre os períodos. Amostras de mamona (Ricinus communis) e carambola (Averrhoa carambola) apresentaram comportamento semelhante. Esses resultados sugerem que a composição química dos extratos pode não variar significativamente entre os períodos de chuva e seca. É importante destacar que um número maior de amostras por período seria necessário para validar essa observação e confirmar essa tendência. O extrato de quebra-pedra apresentou-se como uma amostra diferente das demais, por formar um cluster distinto a esquerda do gráfico de escores, indicando características químicas únicas quando comparado aos outros extratos. O agrupamento distinto observado para Phyllanthus niruri (quebra-pedra) encontra respaldo na literatura. Esta espécie é conhecida por apresentar uma composição rica em lignanas, taninos hidrolisáveis, compostos fenólicos específicos e, especialmente, altos teores de carboidratos e derivados da celulose. Esses componentes são compatíveis com as bandas observadas nos espectros de FTIR (notadamente em 1012 e 1047 cm-1) e justificam sua separação dos demais extratos pela análise de PCA.25,26 Quanto ao agrupamento com os demais extratos (carambola, caju, abacate, erva-cidreira, graviola, entre outros), este pode ser atribuído à presença comum de flavonoides amplamente distribuídos entre essas espécies, como quercetina, rutina e kaempferol, além de outros metabólitos secundários com estruturas químicas semelhantes. Essa composição química resulta em perfis espectrais mais próximos, refletindo-se na proximidade entre essas amostras no gráfico de escores da PCA.27,28 Outro gráfico importante para análise dos dados de infravermelho é o gráfico de pesos, o qual é usado para detectar as áreas espectrais responsáveis pelos agrupamentos. Os pesos são coeficientes pelos quais as variáveis originais devem ser multiplicadas para obter o componente principal em questão. O valor numérico dos pesos indica a contribuição de cada variável original nas componentes principais (PCs), ou seja, quais variáveis contribuem mais para um agrupamento ou separação de amostras.29 Os gráficos de pesos para PC-1 e PC-2 estão apresentados na Figura 6.
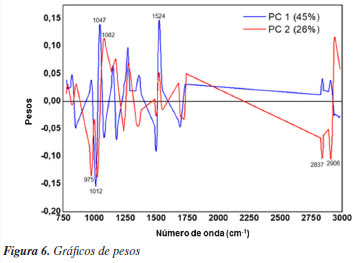
O gráfico de pesos permite identificar as regiões espectrais que contribuem para a separação entre as amostras observadas no gráfico de escores, Figuras 5a e 5b. O PC1 explicou 45% da variação total dos dados e foi responsável por discriminar a amostra de quebra-pedra das demais, apresentando escores negativos no PC1. Essa separação foi influenciada por bandas espectrais mais pronunciadas em 1012 cm-1 (estiramento C-O em carboidratos),30 1047 cm-1 (vibrações de estiramento C-O associadas a derivados de celulose)28 e 1500-1524 cm-1 (vibração de estiramento de C=C em aromáticos).31 Esses resultados estão em concordância com os dados apresentados por Pedro et al.,32 que identificaram a quebra-pedra como rica em carboidratos, com teores variando entre 72,09 e 82,37%. Os pesos associados ao PC2 indicaram bandas mais expressivas em 975, 1024, 1082, 2837, 2906 e 2937 cm-1. As bandas em 975 e 1024 cm-1 foram atribuídas, respectivamente, à vibração de estiramento O-C do grupo O-CH3 presente em polissacarídeos e ao estiramento vibracional O-C do anel heterocíclico central.33,34 Outra absorção relevante foi observada em 1082 cm-1, associada ao estiramento alcoólico de C-O.35 Já na região entre 3000 e 2800 cm-1, as bandas foram atribuídas às vibrações de estiramento de grupos acilas de lipídeos. A diferenciação taxonômica entre as espécies de plantas também foi obtida por análise hierárquica de cluster (HCA), utilizando o método de ligação completa (complete linkage) para o agrupamento, apresentando uma separação semelhante aos resultados apresentados pela PCA. O HCA constrói uma hierarquia de clusters com base na distância Euclidiana e os resultados são apresentados na forma de um dendrograma na Figura 7.
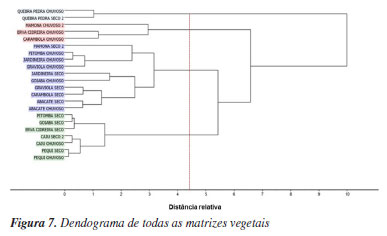
No dendrograma, o eixo x representa a distância relativa entre os clusters, enquanto o eixo y corresponde à representação de todas as amostras. Ao aplicar um corte ao longo da linha vermelha tracejada, observa-se a formação de quatro clusters, em concordância com os resultados obtidos na análise de componentes principais (PCA). A separação dos grupos é evidente, com a amostra de quebra-pedra formando um cluster isolado, semelhante ao comportamento observado na PCA. Esse resultado pode estar relacionado a diferenças específicas na composição química da quebra-pedra, refletidas em uma banda espectral distinta presente em sua estrutura, não apresentada pelas demais matrizes analisadas. Os resultados sugerem que a composição química dos extratos pode não variar significativamente entre os períodos de chuva e seca. É importante destacar que um número maior de amostras por período seria necessário para validar essa observação e confirmar essa tendência. Calibração multivariada Para a análise quantitativa de quercetina, foi utilizada a regressão por mínimos quadrados parciais (PLS), considerando a faixa espectral de 1400-1670 cm-1, localizada na região de impressão digital da molécula de quercetina. Um total de 26 amostras foram analisadas e quantificadas por FTIR associado à quimiometria, das quais 9 amostras foram utilizadas para a validação do modelo. Os espectros foram submetidos a diferentes etapas de pré-processamento, incluindo suavização, aplicação da primeira derivada com filtro Savitzky-Golay (polinômio de 2º grau e janelas de 7 pontos) e centralização dos dados na média. O modelo foi construído com seis variáveis latentes, selecionadas por validação cruzada do tipo leave-one-out (k = 1). Os resultados dos parâmetros estatísticos, como coeficiente de correlação (R), coeficiente de determinação (R2), erro quadrático médio da calibração (RMSEC), erro quadrático médio da validação cruzada (RMSECV) e desvio padrão residual (RPD), para o espectro total e segmentado, são apresentados na Tabela 2.
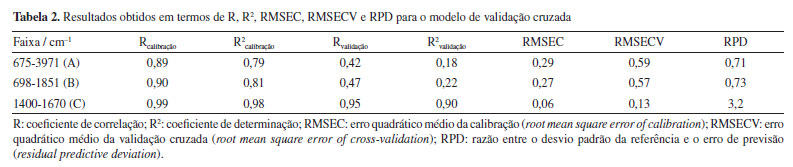
A faixa espectral utilizada para o modelo de calibração, compreendida entre 1400 e 1670 cm-1 (faixa C), apresentou os melhores resultados quando comparada a outras segmentações espectrais. Essa conclusão baseia-se na avaliação do coeficiente de determinação (R2), da raiz do erro médio quadrático, tanto para calibração quanto para validação cruzada (RMSEC e RMSECV), e do desvio padrão residual (RPD). O R2 mede a proporção da variabilidade da variável dependente (Y) explicada pela variável independente (X). De acordo com Karoui et al.,36 valores de R2 entre 0,50 e 0,65 indicam que mais de 50% da variância em Y é explicada por X, representando uma explicação limitada. Valores entre 0,66 e 0,81 indicam previsões quantitativas moderadas, enquanto valores entre 0,82 e 0,90 refletem uma boa previsão. Modelos com R2 superiores a 0,91 são considerados excelentes. Os altos valores de R2, aliados aos baixos valores de RMSEC e RMSECV, demonstram que o modelo de calibração PLS desenvolvido é capaz de prever a concentração de flavonoides. Após a construção e seleção do modelo de calibração mais adequado, foi realizada a validação externa utilizando amostras independentes, que não foram incluídas no conjunto de calibração. A robustez do modelo foi avaliada com base nos valores de R2, RMSEP e RPD, conforme apresentado na Tabela 3. Os dados apresentados nesta tabela foram extraídos do gráfico exibido na Figura 8.
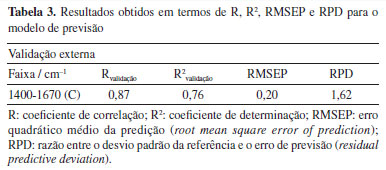
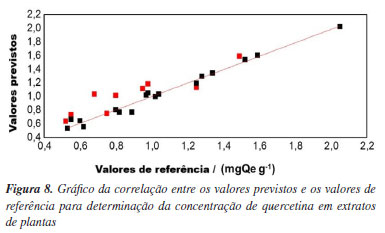
Os poucos trabalhos disponíveis na literatura que determinaram quercetina em plantas medicinais utilizando infravermelho combinado à quimiometria apresentaram erros quadráticos médios superiores aos observados no presente estudo, embora tenham utilizado um número maior de amostras. Camacho-Tamayo et al.37 relataram que o aumento no número de amostras melhora a robustez dos modelos, refletindo em valores mais elevados de R2 e RPD, além de uma redução no RMSE. Isso evidencia que um conjunto maior de amostras permite a criação de modelos mais robustos. O índice RPD avalia a precisão da previsão em relação à variabilidade média das amostras. Segundo Williams e Norris38 um modelo de calibração é considerado satisfatório com valores de RPD entre 1,5 e 2,4, sendo valores acima de 2,4 indicativos de um bom modelo, enquanto valores abaixo de 1,5 sugerem que o modelo não deve ser utilizado. Ao comparar os dados das Tabelas 2 e 3, observa-se uma redução nos valores de Rvalidação, R2validação e RPD, o que já era esperado. Modelos calibrados tendem a apresentar desempenho superior com os dados de calibração em relação aos dados de validação externa, devido à ausência de dados que não participaram de momento algum do processo de ajuste. Além disso, a combinação de amostras com características químicas variadas pode introduzir relações não lineares entre os espectros (X) e as variáveis de interesse (Y), o que representa um desafio adicional para a construção de modelos mais robustos. Como o RMSEP foi maior que o RMSECV, considerou-se a possibilidade de sobreajuste do modelo. Para investigar essa hipótese, foi realizado um teste F comparando as variâncias das amostras de validação em relação ao espectro médio do conjunto de calibração. O valor de F calculado foi 1,82, enquanto o valor crítico da tabela para um nível de confiança de 95% foi 2,59. Como Fcalculado < Fcrítico, conclui-se que o modelo não apresenta indícios de sobreajuste.
CONCLUSÕES O presente trabalho sugere que é possível utilizar a espectroscopia ATR-FTIR associada à quimiometria como metodologia para a determinação da concentração de flavonoides em plantas medicinais. Três agrupamentos foram identificados, os quais estão associados às características químicas distintas dessas amostras, com destaque para o Phyllanthus niruri (quebra-pedra), que se apresentou como a espécie mais discriminada, possivelmente devido às particularidades químicas dessa planta, como o alto teor de carboidratos em sua composição. Os melhores modelos foram obtidos após o tratamento dos espectros com a primeira derivada, evidenciando que a escolha do método de processamento tem um impacto significativo na capacidade de predição do modelo, sendo fundamental a seleção apropriada do método de processamento. Além disso, a faixa espectral também influencia a robustez do modelo. Das três faixas espectrais estudadas, a que apresentou os melhores valores de predição foi a faixa de 1400 a 1670 cm-1, possivelmente devido à sua sensibilidade a vibrações específicas relacionadas aos flavonoides, o que contribui para a exatidão dos modelos.
DECLARAÇÃO DE DISPONIBILIDADE DE DADOS Todos os dados gerados ou analisados durante este estudo estão incluídos neste artigo.
AGRADECIMENTOS O presente trabalho foi realizado com o apoio da Fundação de Amparo à Pesquisa e Desenvolvimento Científico e Tecnológico do Maranhão (FAPEMA). Os autores também agradecem ao Programa de Pós-Graduação em Química (PPGQ/IFMA), ao Laboratório Grupo de Pesquisas, Plantas e Microrganismos, ao Programa de Pós-Graduação em Biodiversidade e Conservação da Universidade Federal do Maranhão representado pelo herbário que prontamente auxiliou na identificação das matrizes vegetais, também agradecem aos professores responsáveis pelo Laboratório de Análise e Pesquisa de Alimentos, Bebidas e Ambiental (LAPAA), que disponibilizaram o ambiente para o desenvolvimento da pesquisa.
CONTRIBUIÇÕES DO AUTOR Nadia B. da Costa foi responsável por conceituação, curadoria de dados, investigação, metodologia, visualização, redação de rascunho original; Matheus F. L. de Oliveira por redação (revisão e edição); Eduardo B. de Almeida Júnior por metodologia; Antônio J. Cantanhede Filho por recursos; Gilmar S. da Silva por análise formal, administração de projetos, aquisição de financiamento; Eliane R. de Sousa por conceituação, análise formal, administração de projetos, metodologia, recursos, software, validação, redação (revisão e edição). Todos os autores leram e aprovaram a versão final do manuscrito.
REFERÊNCIAS 1. Kapadia, P.; Newell, A. S.; Cunningham, J.; Roberts, M. R.; Hardy, J. G.; Int. J. Mol. Sci. 2022, 23, 10334. [Crossref] 2. Seidel, V.; Plants 2020, 9, 1562. [Crossref] 3. Matsubara, Y.; Yuki Gosei Kagaku Kyokaishi 1992, 50, 963. [Crossref] 4. Wulandari, L.; Retnaningtyas, Y.; Nuri; Lukman, H.; J. Anal. Methods Chem. 2016, 2016, 4696803. [Crossref] 5. Wulandari, L.; Permana, B. D.; Kristiningrum, N.; Indones. J. Chem. 2020, 20, 1230. [Crossref] 6. Siddiqui, M. R.; AlOthman, Z. A.; Rahman, N.; Arab. J. Chem. 2017, 10, S1409. [Crossref] 7. Johnson, J. B.; Walsh, K. B.; Naiker, M.; Ameer, K.; Molecules 2023, 28, 3215. [Crossref] 8. Bureau, S.; Cozzolino, D.; Clark, C. J.; Postharvest Biol. Technol. 2019, 148, 10. [Crossref] 9. Zou, L.; Li, H.; Ding, X.; Liu, Z.; He, D.; Kowah, J. A. H.; Wang, L.; Yuan, M.; Liu, X.; Molecules 2022, 27, 77766. [Crossref] 10. de Oliveira, E. C.; Brazilian Journal of Analytical Chemistry 2025, 12, 11. [Crossref] 11. Li, Y.; Shen, Y.; Yao, C. L.; Guo, D.; J. Pharm. Biomed. Anal. 2020, 185, 113215. [Crossref] 12. Oskolkov, N. Em Applied Data Science in Tourism; Egger, R., ed.; Springer: Cham, 2022, p. 169. [Crossref] 13. Ferreira, M. M. C.; Quimiometria: Conceitos, Métodos e Aplicações, 1ª ed.; Editora da Unicamp: Campinas, 2015. 14. Liu, J.; Wena, Y.; Dong, N.; Lai, C.; Zhao, G.; Food Chem. 2013, 141, 3977. [Crossref] 15. Liu, X.; Jiang, W.; Su, M.; Sun, Y.; Liu, H.; Nie, L.; Zang, H.; J. Sep. Sci. 2020, 43, 6. [Crossref] 16. Popa, R. M.; Fetea, F.; Socaciu, C.; Molecules 2021, 26, 4837. [Crossref] 17. Sierra, P. M. L.; Gallardo-Velázquez, T.; Osorio-Revilla, G.; Meza-Márquez, O. G.; Cienc. Tecnol. Aliment. 2019, 17, 724. [Crossref] 18. Gordon, R.; Chapman, J.; Power, A.; Chandra, S.; Roberts, J.; Cozzolino, D.; J. Cereal Sci. 2019, 85, 41. [Crossref] 19. Maharaj, A.; Naidoo, Y.; Dewir, Y. H.; Rihan, H.; Horticulturae 2022, 8, 909. [Crossref] 20. Zilli, G. A. L.; Locateli, G.; Zanotelli, P.; Zanetti, M.; Zeferino, R. C. F.; Fiori, M. A.; Gasparetto, A.; Anais do 9º Seminário Integrado de Ensino, Pesquisa e Extensão da Unochapecó; Chapecó, Brasil, 2019. [Link] acessado em agosto 2024 21. Pourcel, L.; Routaboul, J. M.; Cheynier, V.; Lepiniec, L.; Debeaujon, I.; Trends Plant Sci. 2007, 12, 29. [Crossref] 22. Szymanska-Chargot, M.; Zdunek, A.; Food Biophysics 2013, 8, 29. [Crossref] 23. Caunii, A.; Pribac, G.; Grozea, I.; Gaitin, D.; Samfira, I.; Chem. Cent. J. 2012, 6, 123. [Crossref] 24. de Souza, A. M.; Poppi, R. J.; Quim. Nova 2012, 35, 152. [Crossref] 25. Pius, O.; Pius, A. O.; Babawale, P. O.; Adebimpe, E.; Sola, A.; Comfort, O.; J. Toxicol. Sci. 2015, 7, 255. [Crossref] 26. Calixto, J. B.; Santos, A. R. S.; Cechinel Filho, V.; Yunes, R. A.; Med. Res. Rev. 1998, 18, 225. [Crossref] 27. Baptista, A.; Gonçalves, R. V.; Bressan, J.; Pelúzio, M. C. G.; Oxid. Med. Cell. Longevity 2018, 2018, 3753562. [Crossref] 28. Heneczkowski, M.; Kopacz, M.; Nowak, D.; Kuźniar, A.; Acta Poloniae Pharmaceutica - Drug Research 2001, 58, 415. [Link] acessado em Agosto 2025 29. Hssaini, L.; Razouk, R.; Bouslihim, Y.; Front. Plant Sci. 2022, 13, 782159. [Crossref] 30. Sousa, C. M. M.; Silva, H. R.; Vieira Junior, G. M.; Ayres, M. C. C.; da Costa, C. S. L.; Araújo, D. S.; Cavalcante, L. D. C.; Barros, E. S. D.; Araújo, P. M. B.; Brandão, M. S.; Chaves, M. H.; Quim. Nova 2007, 30, 1199. [Crossref] 31. Dharmaraj, S.; Jamaludin, A. S.; Razak, H. M.; Valliappan, R.; Ahmad, N. A.; Harn, G. L.; Ismail, Z.; Vib. Spectrosc. 2006, 41, 68. [Crossref] 32. Karabagias, I. K.; Foods 2020, 9, 1550. [Crossref] 33. Hammes, N.; Pinheiro, C.; Segundo, I. R.; Homem, N. C.; Silva, M. M.; Felgueiras, H. P.; Soares, G. M. B.; Freitas, E.; Costa, M. F. M.; Carneiro, J. A. O.; Appl. Sci. 2024, 14, 2473. [Crossref] 32. Pedro, F. G. G.; Arruda, G. L.; Oliveira, J. C.; Santos, A. D.; Sigarini, K. S.; Hernandes, T.; Villa, R. D.; Oliveira, A. P.; Rev. Bras. Plant. Med. 2016, 18, 297. [Crossref] 35. Krysa, M.; Szymańska-Chargot, M.; Zdunek, A.; Food Chem. 2022, 393, 133430. [Crossref] 36. Karoui, R.; Mouazen, A. M.; Dufour, E.; Pillonel, L.; Schaller, E.; De Baerdemaeker, J.; Bosset, J. O.; Int. Dairy J. 2006, 16, 1211. [Crossref] 37. Camacho-Tamayo, J. H.; Rubiano S. Y.; Hurtado, S. M. P.; Agronomia Colombiana 2014, 32, 86. [Crossref] 38. Williams, P.; Norris, K.; Mol. Nutr. Food Res. 1988, 32, 803. [Crossref]
Editor Associado responsável pelo artigo: Eduardo M. Richter |

On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access